Acknowledgments:
Laboratory of Chemical Physics


Laboratory of Biochemistry and Genetics


*Support provided by the Intramural Research Division of the NIDDK, NIH.
Protein Structure
Primary structure (sequence)
GSIGAASMEF CFDVFKELKV HHANENIFYC PIAIMSALAM VYLGAKDSTR TQINKVVRFD KLPGFGDEIE AQCGTSVNVH
SSLRDILNQI TKPNDVYSFS LASRLYAEER YPILPEYLQC VKELYRGGLE PINFQTAADQ ARELINSWVE SQTNGIIRNV
LQPSSVDSQT AMVLVNAIVF KGLWEKAFKD EDTQAMPFRV TEQESKPVQM MYQIGLFRVA SMASEKMKIL ELPFASGTMS
MLVLLPDEVS GLEQLESIIN FEKLTEWTSS NVMEERKIKV YLPRMKMEEK YNLTSVLMAM GITDVFSSSA NLSGISSAES
LKISQAVHAA HAEINEAGRE VVGGAEAGVD AASVSEEFRA DHPFLFCIKH IATNAVLFFG RCVSP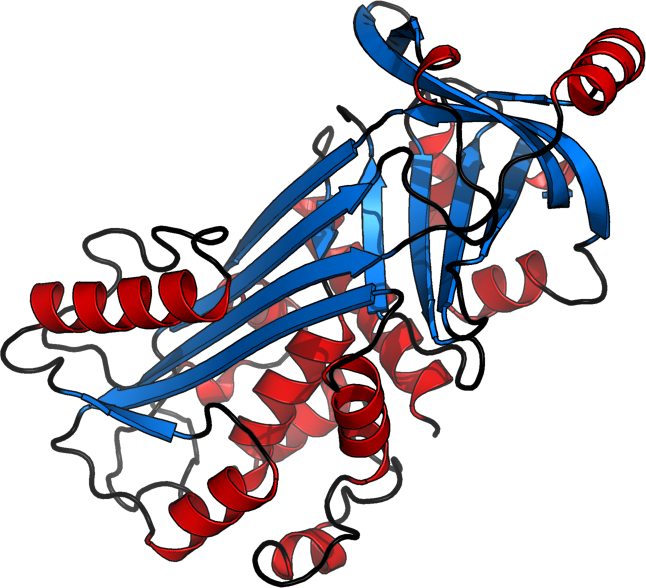
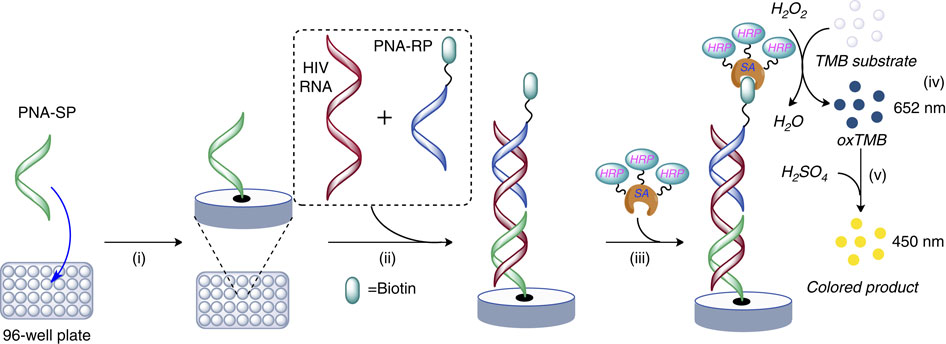
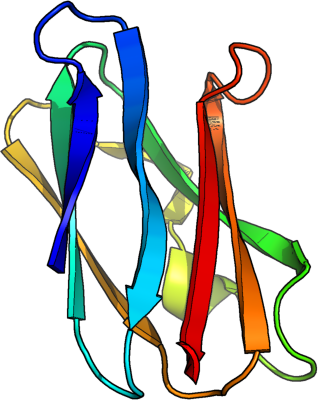
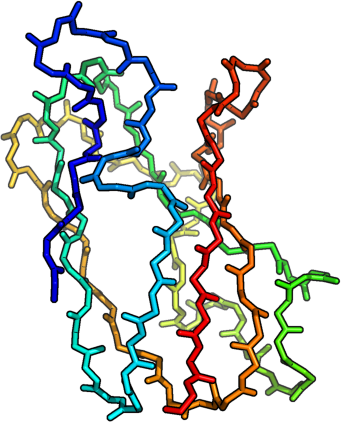
helices [red], sheets [blue
3D structure
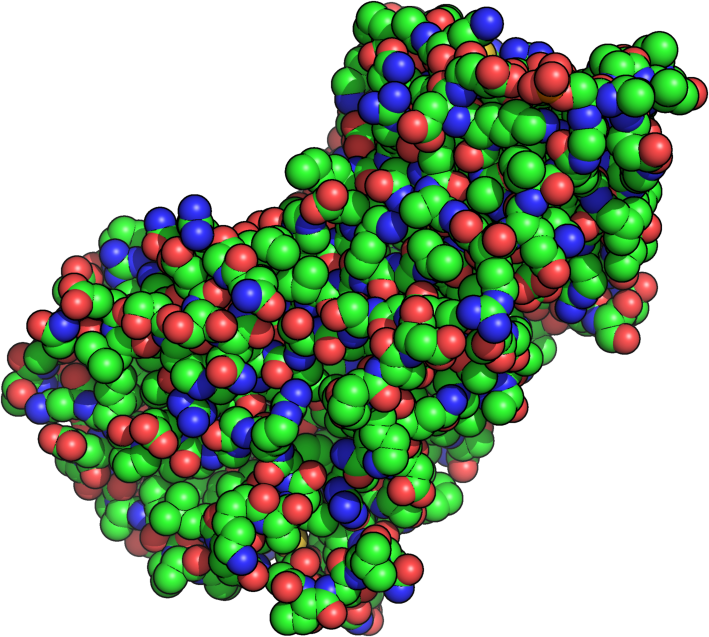
complexes, aggregation
SEM Aggregate structure, Zabik et al., J. Poul. Sci. (1980)
Primary Structure
Twenty residue "alphabet" forms polypeptide chain
Protein folding problem
Predict structure from sequence
Sequence Structure Function
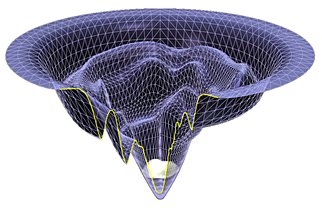
Native structure, folding pathways, ...
MD simulation of WW-domain, Best and Mittal, J. Phys. Chem. B (2010)
Scientific Philosophy
Theoreticians need to keep in close contact with experimentalists.Imagination must be constrained by reality.


Higher order structure
Phase separations lead to sudden changes in liquid structure.

How do we model many protein-protein interactions?
Can we predict aggregates from experimental structure?
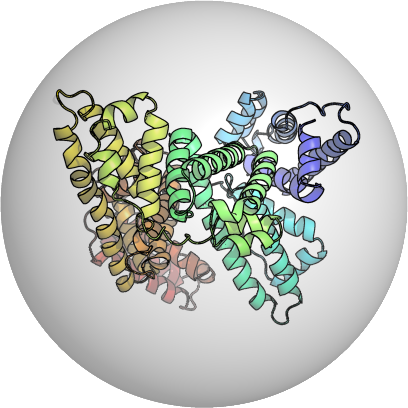
PDB:1AO6
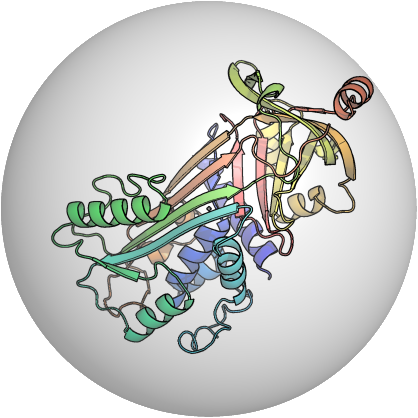
PDB:1OVA

PDB:1W6Z
PDB:3V03
Experimental Measurements
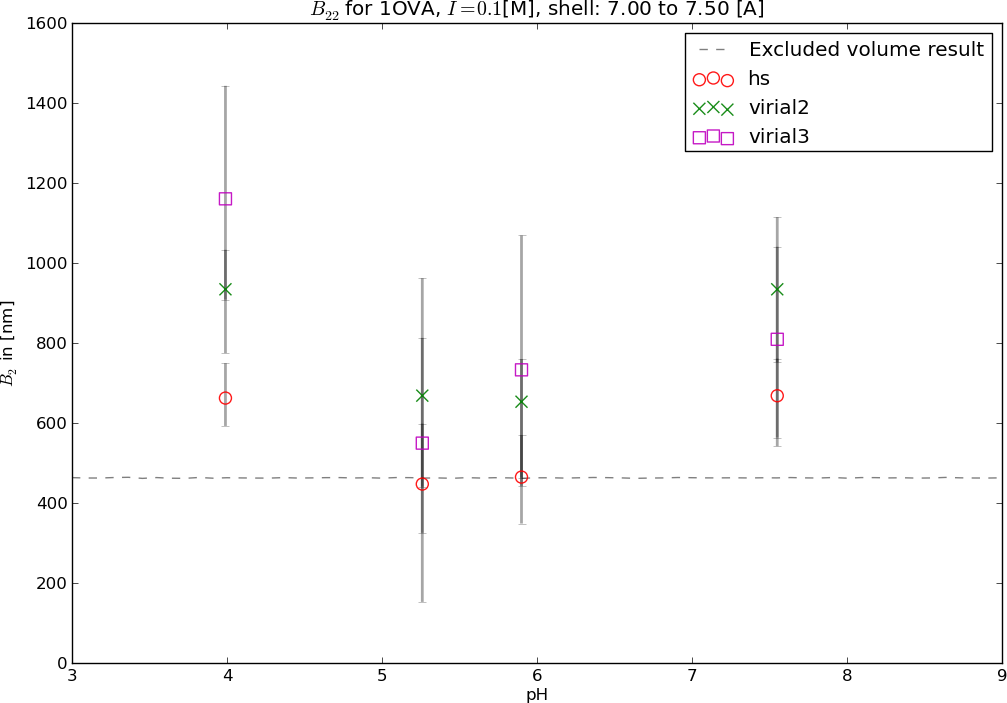
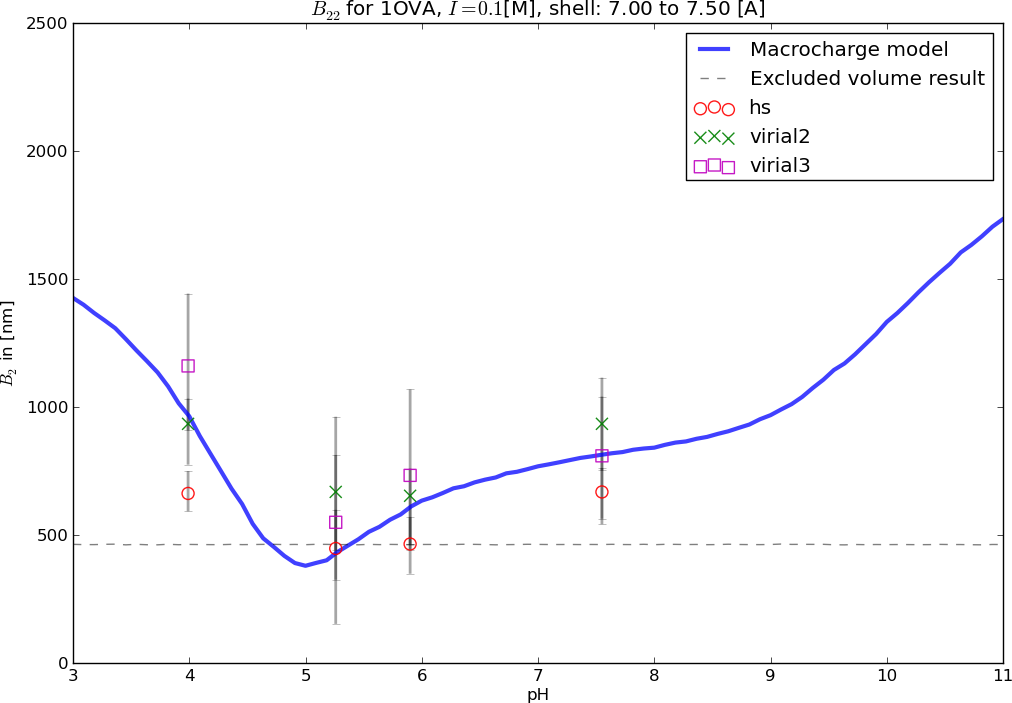
Second virial coefficient, , measurementusing light scattering at different pH.
The Process

Start with the crystallized PDB Structure
e.g. Human Serum Albumin PDB:1A06
Electrostatics: Poisson-Boltzmann
Solve for with the Adaptive Poisson-Boltzmann Solver (APBS),
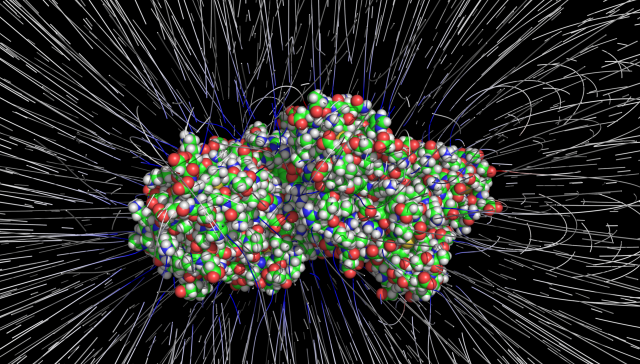
Typically (in the absence of ions), and .
Macrocharge fitting
Best fit macrocharges to approximate the field.
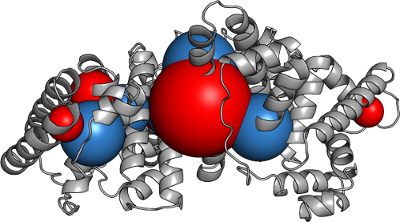
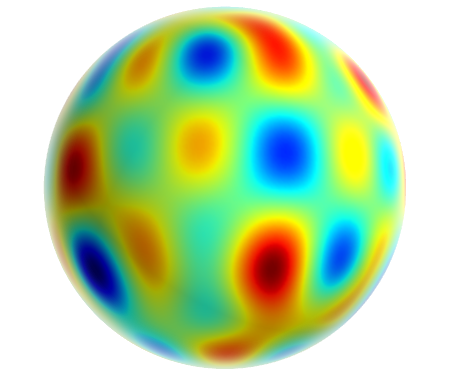
Decompose the field, determine a region of excluded volume +
spherical harmonic decomposition for large distances.
Matching experiments
Theoretical predictions of the second virial coefficientconsidering only excluded volume and reduced electrostatics.
Matching experiments
Theoretical predictions of the second virial coefficientconsidering only excluded volume and reduced electrostatics.
Paradigm shift
Proteins were thought to adopt stable, folded conformations.Solving the structure was paramount for understanding the function.
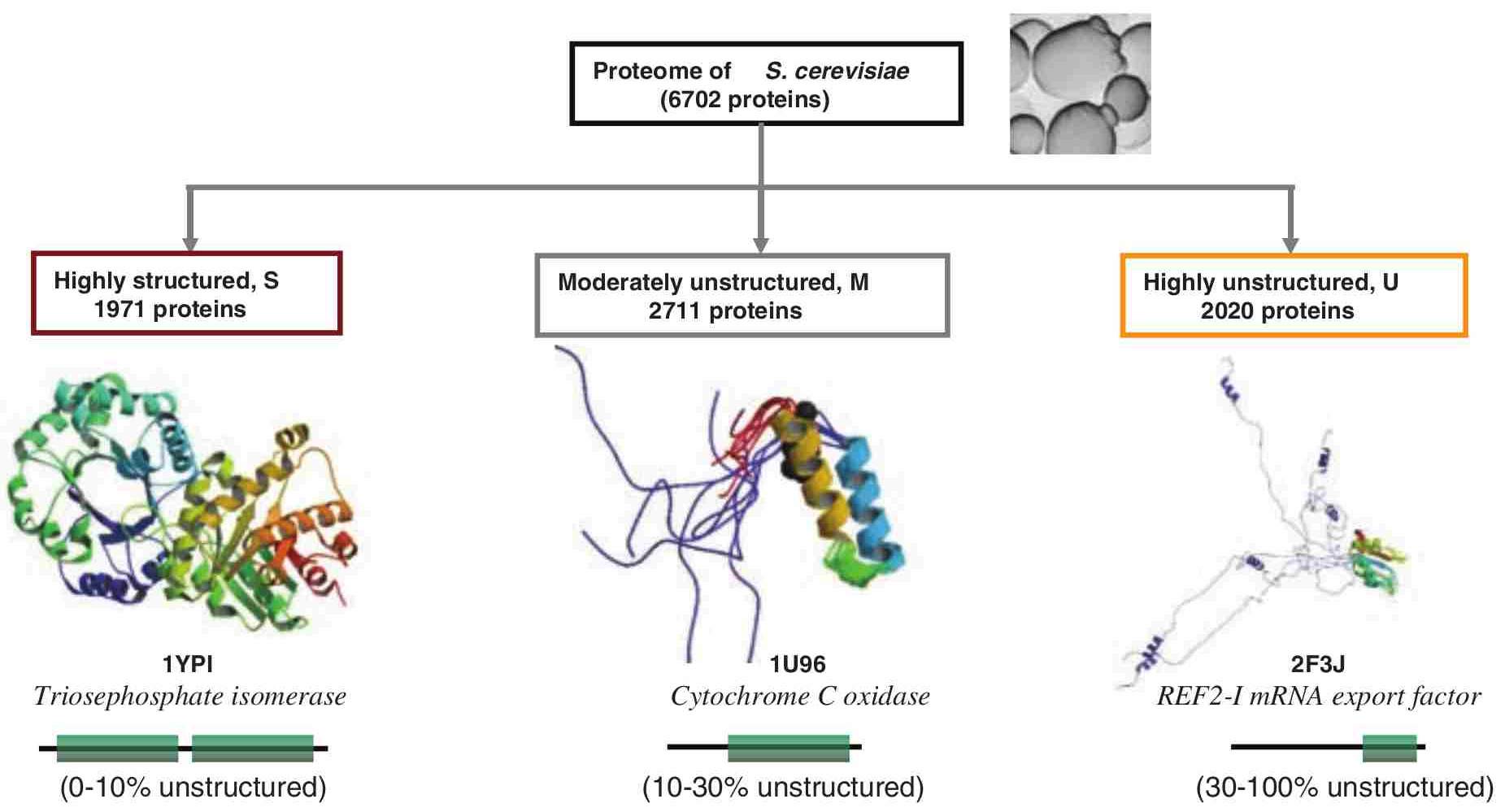
Intrinsically disordered proteins
Structure
- Lack tertiary structure (disorder!)
- Still may form secondary structure
- Different primary structure (residue propensity)
- More charged, less hydrophobic and aromatic residues
Binding
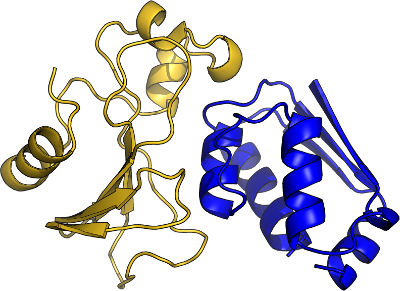
Barnase-Barstar complex

Hif-1 α/CBP
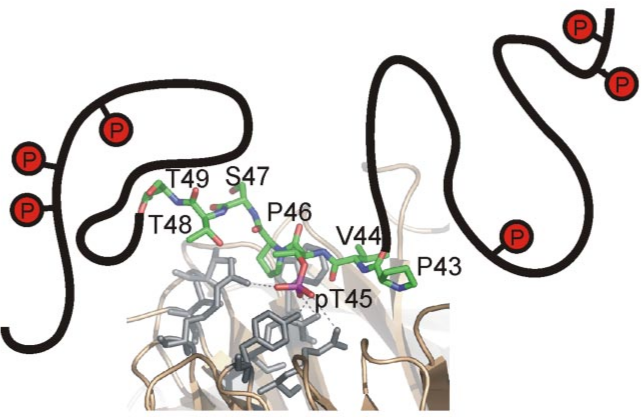
SIC1 binding to CDC4
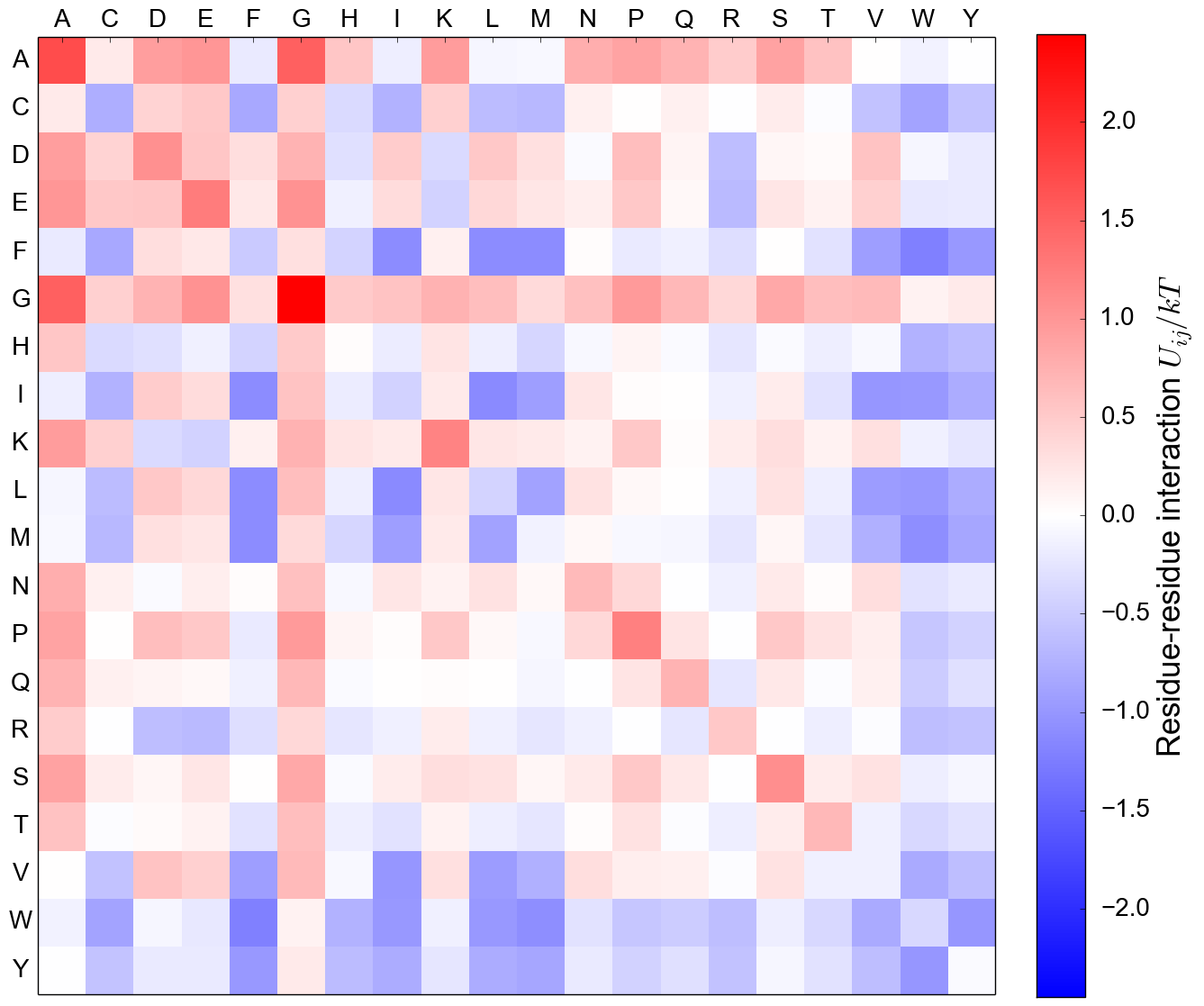
Statistical Potentials
Residue-residue interactions, quasi-chemical lattice-gas

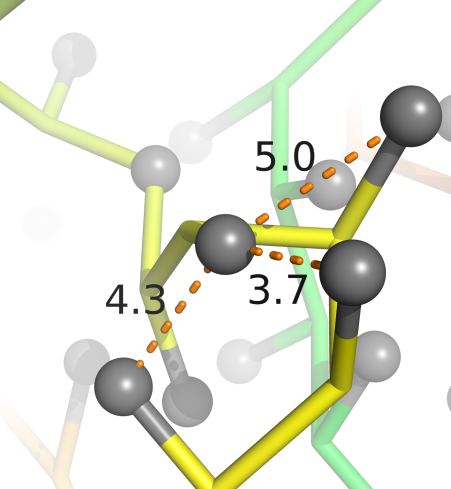
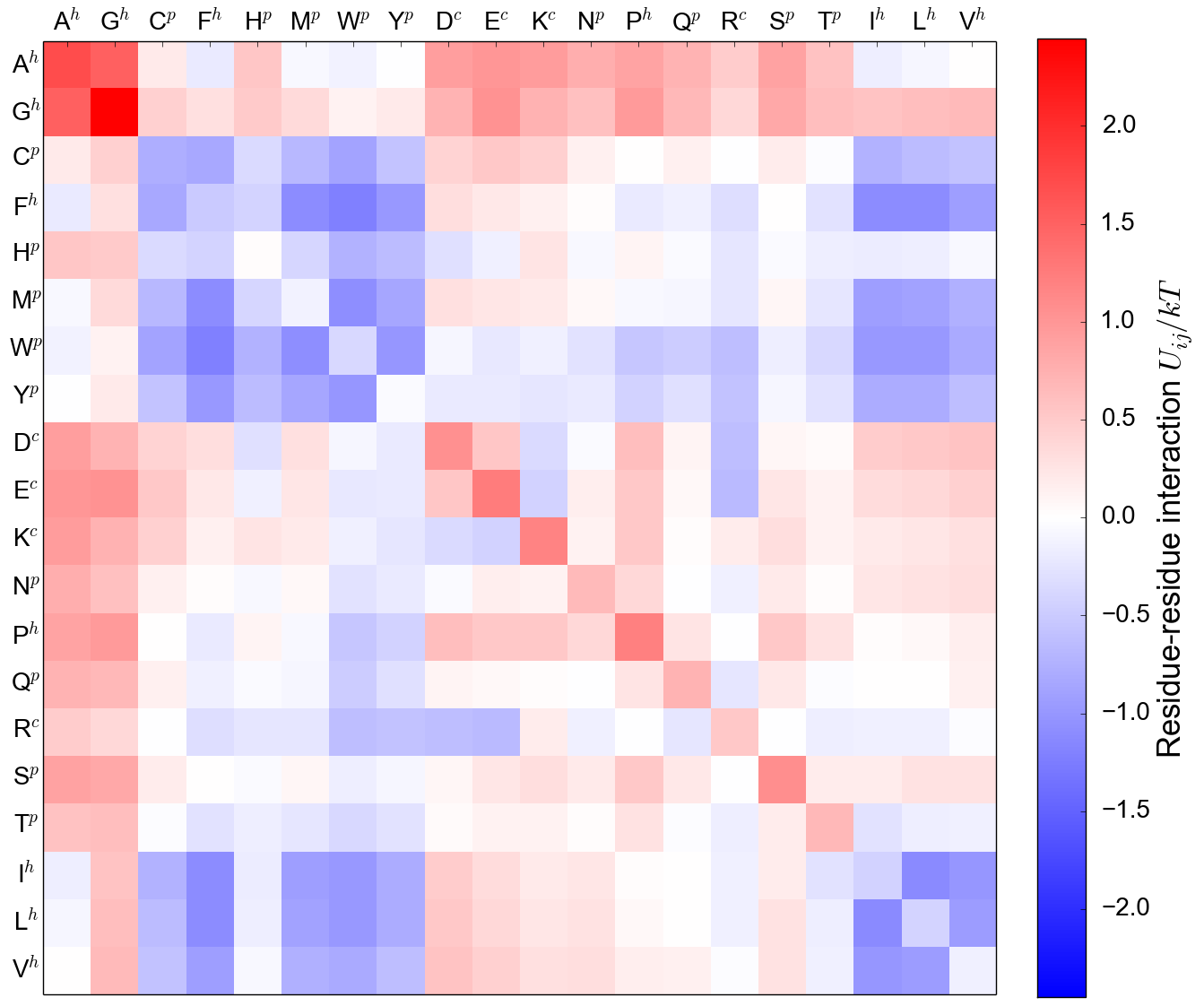
Residue-residue interaction matrix, MJ
Betancourt and Thirumalai (1999), Skolnick, Kolinski and Ortiz (2000)
MJ matrix reveals biophysical structure
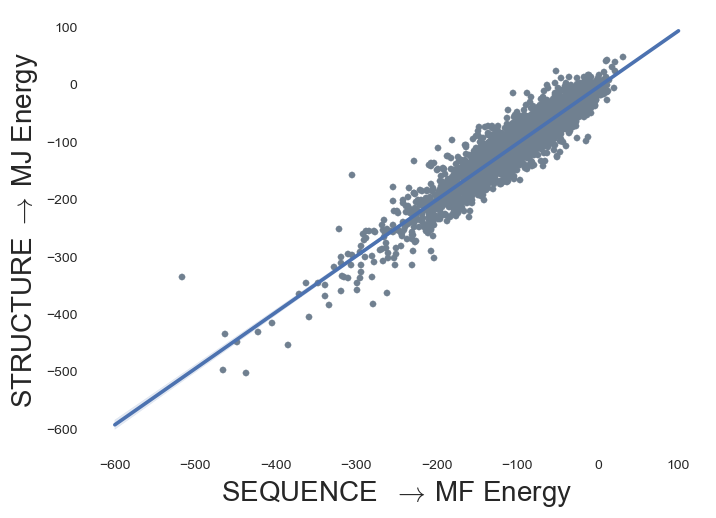
MJ contact energy reproduces MF energy
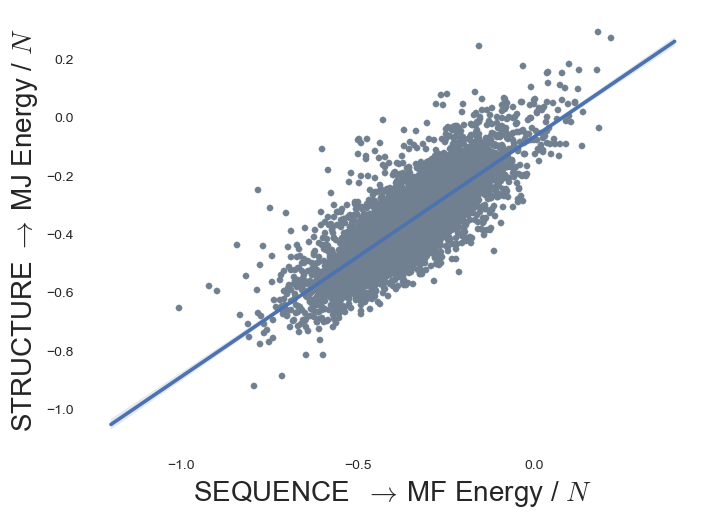{kind=link}
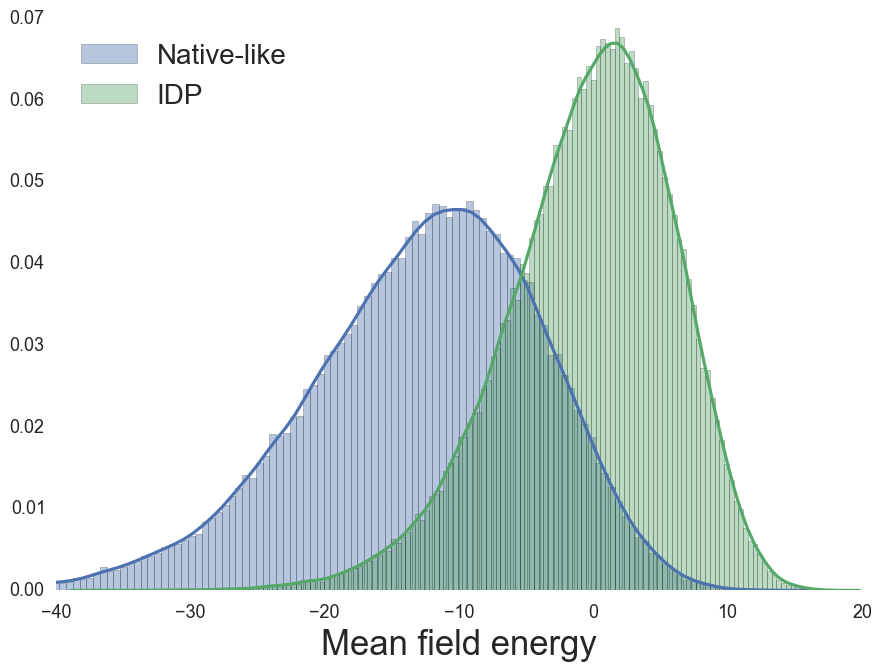
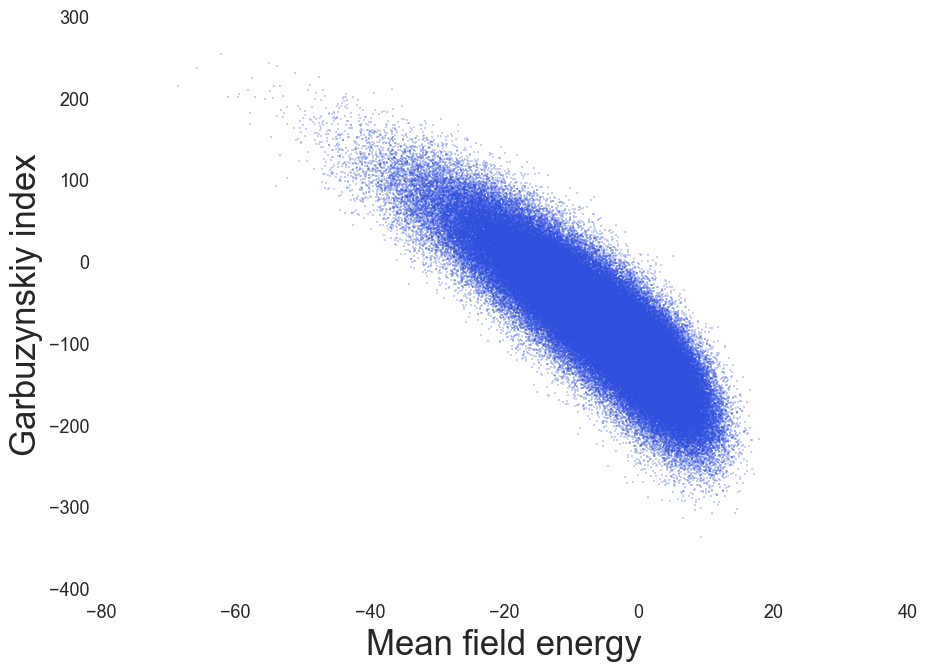
MF Energy distributions: Physically reasonable

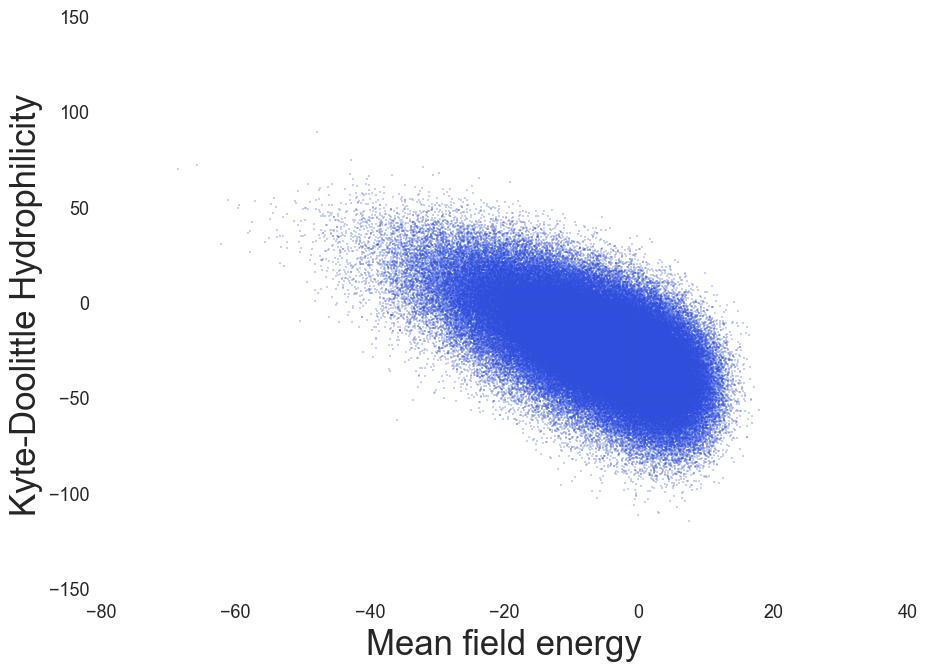
Hydrophilicity index, Kyte & Doolittle, J. Mol. Biol. (1982)
Amyloidogenic regions, Garbuzynskiy et. al. Bioinformatics (2010)
Protein Networks
- Target protein interacts with a range of possible surfaces.
- Measure average binding affinity of protein to surfaces.
- Measure binding specificity of protein to surfaces.

Schwikowski & Fields et al., Nature 2000.
Binding affinity

Binding specificity
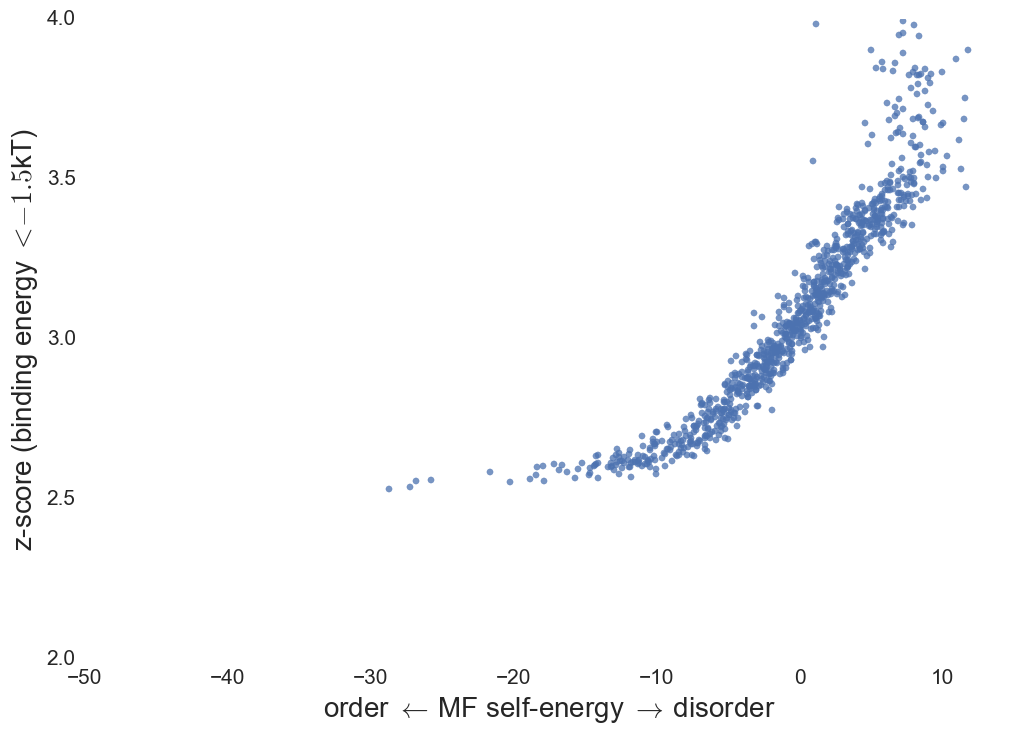
MF IDP Summary:
- MF models reproduce MJ contact energies. MF IDP's bound to native structures show increased specificity with lower affinity.


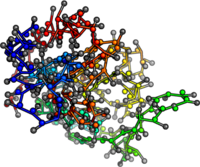
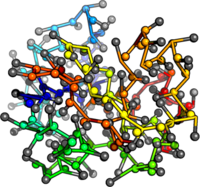



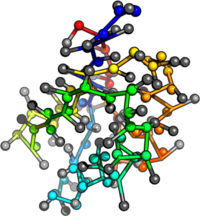

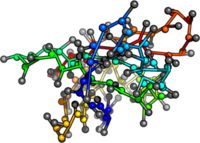
What's next? Add structure to mean field calculations.
Lattices may be optimal for IDP's, they can reproduce native-energies but quickly sample extended conformational space.
Active research projects & collaborations
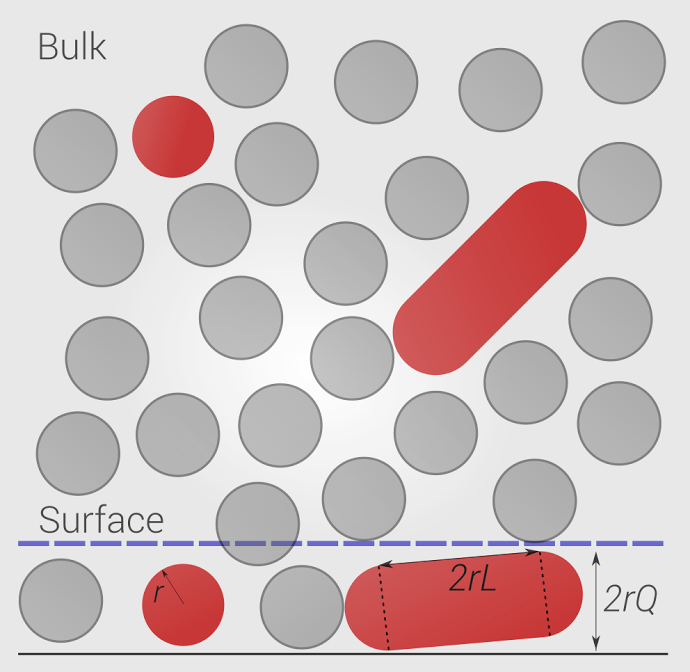
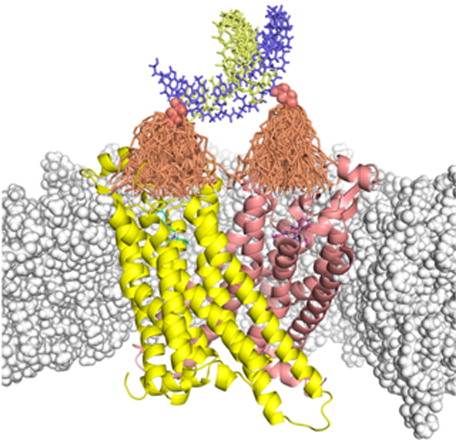
Crowding, surface adsorption and protein fibrillation, Biophys (in press).Programmable Nanoscaffolds and Multivalent Effects, JACS.
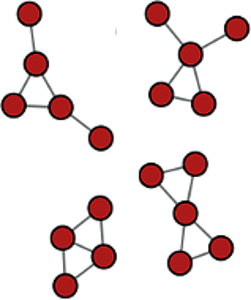
Integer sequence discovery from small graphs, Discrete Math. (submitted).
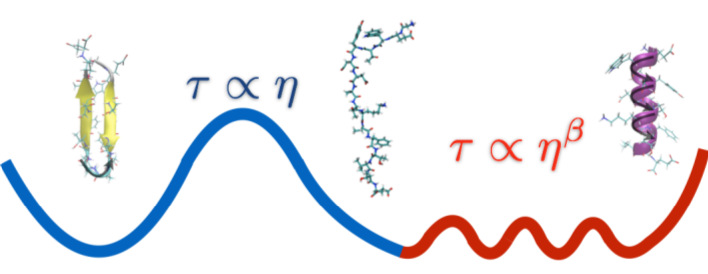
Dependence of Internal Friction on Folding Mechanism, JACS (submitted).
Quantification of plasma HIV RNA, Nature Comm.