intra-protein interactions

Human serum albumin, Ovalbumin, Lysozyme, Bovine Serum Albumin respectively.
How do we model the interaction between proteins on a larger scale? Can we predict aggregates?
Mixtures
Phase separations lead to sudden fundamental changes in liquid structure and local density.


This is usually really important.
Virial Coefficients
Why work with this expansion?Experimental measurements (light scattering) possible.
The Process

Start with the crystallized PDB Structure (HSA)
Adaptive Poisson-Boltzmann Solver
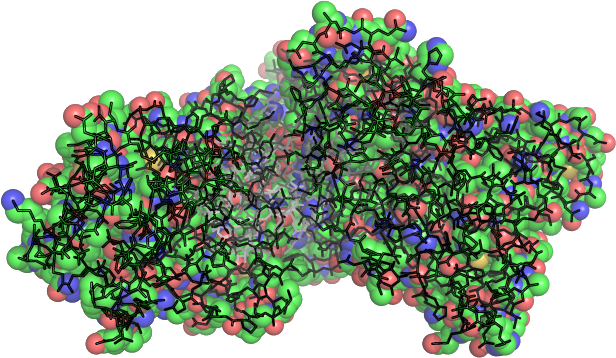
Typically (in the absence of ions)
,
The Process
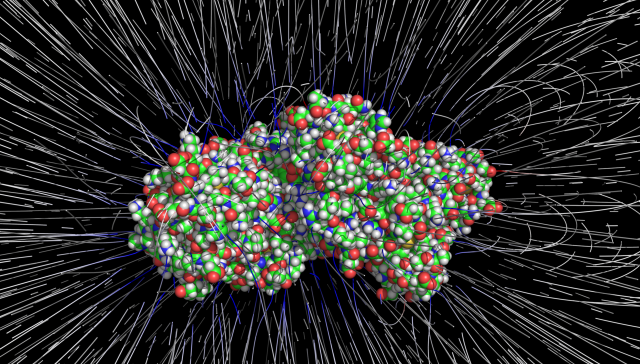
Electrostatic field
The Process
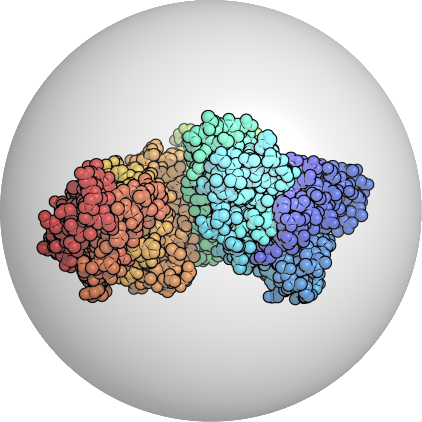
Determine a region of excluded volume.
The Process
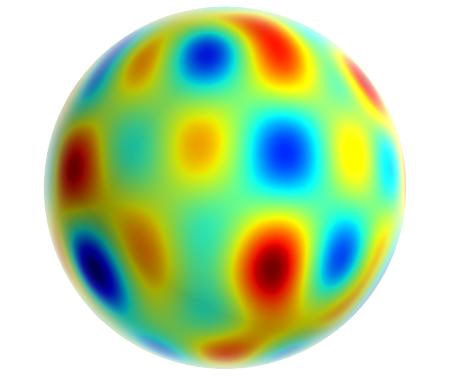
Spherical Harmonic decomposition for large distances.
The Process
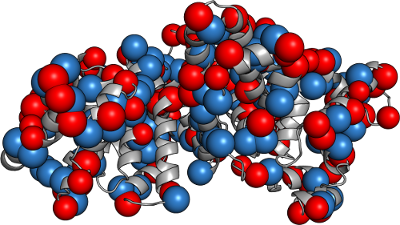
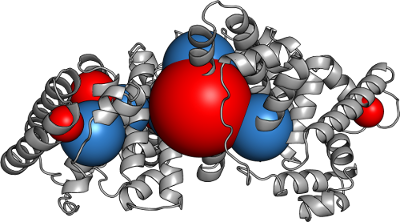
Best fit macrocharges to replicate the field.
This works

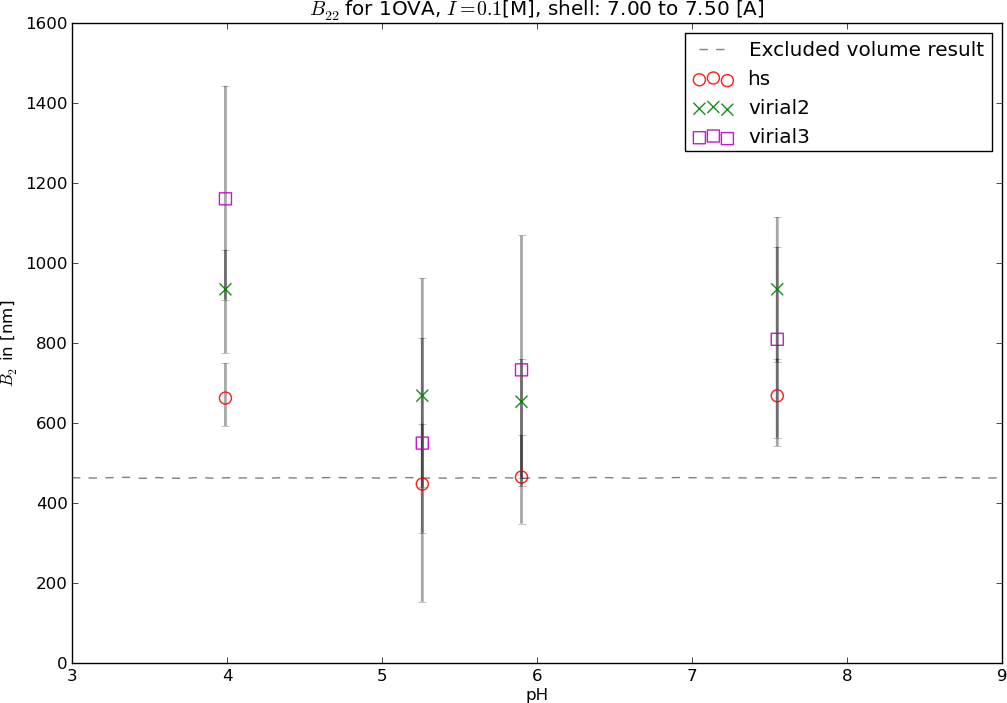
Results for Ovalbumin

Future work
Large scale Monte-Carlo simulation using macrocharges.

results for square-well potentials shown